
Abstract
Patients with severe COVID-19 have a hyperinflammatory immune response suggestive of macrophage activation. Bruton tyrosine kinase (BTK) regulates macrophage signaling and activation. Acalabrutinib, a selective BTK inhibitor, was administered off-label to 19 patients hospitalized with severe COVID-19 (11 on supplemental oxygen; 8 on mechanical ventilation), 18 of whom had increasing oxygen requirements at baseline. Over a 10-14 day treatment course, acalabrutinib improved oxygenation in a majority of patients, often within 1-3 days, and had no discernable toxicity. Measures of inflammation – C-reactive protein and IL-6 – normalized quickly in most patients, as did lymphopenia, in correlation with improved oxygenation. At the end of acalabrutinib treatment, 8/11 (72.7%) patients in the supplemental oxygen cohort had been discharged on room air, and 4/8 (50%) patients in the mechanical ventilation cohort had been successfully extubated, with 2/8 (25%) discharged on room air. Ex vivo analysis revealed significantly elevated BTK activity, as evidenced by autophosphorylation, and increased IL-6 production in blood monocytes from patients with severe COVID-19 compared with blood monocytes from healthy volunteers. These results suggest that targeting excessive host inflammation with a BTK inhibitor is a therapeutic strategy in severe COVID-19 and has led to a confirmatory international prospective randomized controlled clinical trial.
INTRODUCTION
Coronavirus 2019 (COVID-19) is a new pandemic disease caused by a single-stranded RNA zoonotic virus termed severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (1). The spectrum of COVID-19 ranges from a mild respiratory illness to a severe disease requiring hospitalization in up to a third of patients, with frequent progression to acute respiratory distress syndrome (ARDS) and a high mortality (2). It has been reported that COVID-19 patients can have a biphasic clinical course with deterioration following initial improvement, consistent with a delayed and exaggerated immune activation (2–4). A virus-induced hyperinflammatory response or “cytokine storm” (5) has been hypothesized to be a major pathogenic mechanism of ARDS in these patients through modulation of pulmonary macrophages, dendritic cells and/or neutrophils (6–10). Indeed, patients with COVID-19 have elevated blood levels of multiple inflammatory cytokines and chemokines (IL-1β, IL-6, IL-7, IL-8, IL-9, IL-10, G-CSF, GM-CSF, IFN-γ, IP-10, MCP-1, and MIP-1α), and those requiring admittance to an intensive care unit have even higher levels of many of these (11, 12). The hyperinflammatory response in COVID-19 shares biological characteristics with macrophage activation syndrome, suggesting that targeting the innate immune system may be an effective strategy (13).
We became aware of the role of Bruton tyrosine kinase (BTK) in human innate immune responses from our studies of the BTK inhibitor ibrutinib in lymphoma, in which some patients developed invasive aspergillosis during treatment (14). Moreover, we demonstrated that BTK-deficient mice are unable to control infection with this fungus, which is normally kept in check by monocytes/macrophages and neutrophils (14–16). While this is an uncommon complication, it raised the possibility that BTK inhibitors may modulate human inflammatory responses dominated by macrophages, as is the case in COVID-19 (17, 18) and in a mouse model of this infection (Fig. 1) (19). In macrophages, Toll-like receptors (TLRs) recognize single-stranded RNA from viruses such as SARS-CoV-2 and initiate signaling through BTK-dependent activation of NF-κB, triggering the production of multiple inflammatory cytokines and chemokines as well as phagocytosis (Fig. 1) (20–23). In addition, BTK plays a key role in the activation of the NLRP3 inflammasome, resulting in maturation and secretion of IL-1β (24–26). Moreover, in a mouse influenza model, BTK inhibition decreased inflammatory mediators and rescued mice from lethal acute lung injury, suggesting that it may mitigate virally-induced lung damage driven by excessive inflammation (27).
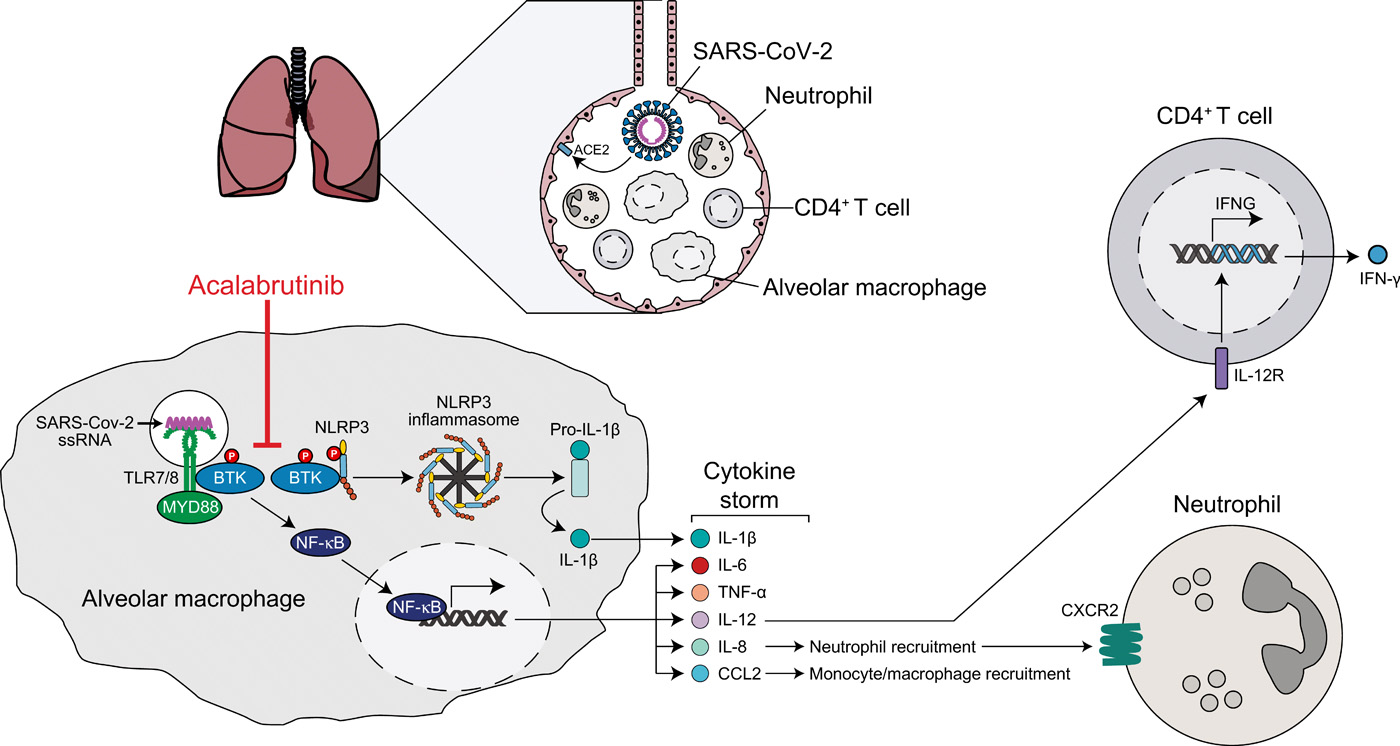
Fig. 1 Model of BTK-dependent hyper-inflammation in severe COVID-19.
Binding of SARS-CoV2 to ACE2 on respiratory epithelia initiates infection. Hypothetically, macrophages may participate in the COVID-19 inflammatory response by phagocytic uptake of viral particles or cellular debris containing viral single-stranded RNA (ssRNA). ssRNA can bind to TLR7 and TLR8, thereby recruiting and activating BTK and MYD88 (51, 52). Downstream of TLR engagement, BTK-dependent NF-κB activation results in the production of pro-inflammatory cytokines and chemokines (53), a “cytokine storm” that could increase the recruitment of monocytes/macrophages and neutrophils during the late phase of severe COVID-19 infection. BTK inhibitors such as acalabrutinib block TLR-dependent NF-κB activation in macrophages (20, 21), thereby dampening the production of pro-inflammatory mediators, as occurs in an influenza-induced lung injury model (27). During severe COVID-19, the heightened levels of IL-1β in several COVID-19 patients (11, 12) indicates the formation of an NLRP3 inflammasome that converts pro-IL-1β to mature IL-1β (54). BTK binds to and phosphorylates NLRP3, thereby promoting its oligomerization and assembly into an inflammasome (24–26). BTK inhibitors such as acalabrutinib inhibit inflammasome-mediated production of IL-1β, as observed in a model of influenza-induced lung injury (27). SARS-CoV-2, severe acute respiratory syndrome coronavirus 2; COVID-19, coronavirus disease 2019; ACE2, angiotensin-converting enzyme 2; TLR, Toll-like receptor; MyD88, myeloid differentiation primary response 88; BTK, Bruton tyrosine kinase; NF-κB, nuclear factor kappa B; NLRP3, NLR family pyrin domain containing 3; ASC, Apoptosis-associated speck-like protein containing a caspase recruitment domain; ORF3a, open reading frame 3a; IFN-γ, interferon gamma; IL, interleukin; IL-12R, IL-12 receptor; CCL2, C-C motif chemokine ligand 2; CXCL1, C-X-C motif chemokine ligand 1; CXCR2, C-X-C motif chemokine receptor 2.
” data-hide-link-title=”0″ data-icon-position=”” href=”https://immunology.sciencemag.org/content/immunology/5/48/eabd0110/F1.large.jpg?width=800&height=600&carousel=1″ rel=”gallery-fragment-images-1266750347″ title=”Model of BTK-dependent hyper-inflammation in severe COVID-19. Binding of SARS-CoV2 to ACE2 on respiratory epithelia initiates infection. Hypothetically, macrophages may participate in the COVID-19 inflammatory response by phagocytic uptake of viral particles or cellular debris containing viral single-stranded RNA (ssRNA). ssRNA can bind to TLR7 and TLR8, thereby recruiting and activating BTK and MYD88 (51, 52). Downstream of TLR engagement, BTK-dependent NF-κB activation results in the production of pro-inflammatory cytokines and chemokines (53), a “cytokine storm” that could increase the recruitment of monocytes/macrophages and neutrophils during the late phase of severe COVID-19 infection. BTK inhibitors such as acalabrutinib block TLR-dependent NF-κB activation in macrophages (20, 21), thereby dampening the production of pro-inflammatory mediators, as occurs in an influenza-induced lung injury model (27). During severe COVID-19, the heightened levels of IL-1β in several COVID-19 patients (11, 12) indicates the formation of an NLRP3 inflammasome that converts pro-IL-1β to mature IL-1β (54). BTK binds to and phosphorylates NLRP3, thereby promoting its oligomerization and assembly into an inflammasome (24–26). BTK inhibitors such as acalabrutinib inhibit inflammasome-mediated production of IL-1β, as observed in a model of influenza-induced lung injury (27). SARS-CoV-2, severe acute respiratory syndrome coronavirus 2; COVID-19, coronavirus disease 2019; ACE2, angiotensin-converting enzyme 2; TLR, Toll-like receptor; MyD88, myeloid differentiation primary response 88; BTK, Bruton tyrosine kinase; NF-κB, nuclear factor kappa B; NLRP3, NLR family pyrin domain containing 3; ASC, Apoptosis-associated speck-like protein containing a caspase recruitment domain; ORF3a, open reading frame 3a; IFN-γ, interferon gamma; IL, interleukin; IL-12R, IL-12 receptor; CCL2, C-C motif chemokine ligand 2; CXCL1, C-X-C motif chemokine ligand 1; CXCR2, C-X-C motif chemokine receptor 2.”>
Binding of SARS-CoV2 to ACE2 on respiratory epithelia initiates infection. Hypothetically, macrophages may participate in the COVID-19 inflammatory response by phagocytic uptake of viral particles or cellular debris containing viral single-stranded RNA (ssRNA). ssRNA can bind to TLR7 and TLR8, thereby recruiting and activating BTK and MYD88 (51, 52). Downstream of TLR engagement, BTK-dependent NF-κB activation results in the production of pro-inflammatory cytokines and chemokines (53), a “cytokine storm” that could increase the recruitment of monocytes/macrophages and neutrophils during the late phase of severe COVID-19 infection. BTK inhibitors such as acalabrutinib block TLR-dependent NF-κB activation in macrophages (20, 21), thereby dampening the production of pro-inflammatory mediators, as occurs in an influenza-induced lung injury model (27). During severe COVID-19, the heightened levels of IL-1β in several COVID-19 patients (11, 12) indicates the formation of an NLRP3 inflammasome that converts pro-IL-1β to mature IL-1β (54). BTK binds to and phosphorylates NLRP3, thereby promoting its oligomerization and assembly into an inflammasome (24–26). BTK inhibitors such as acalabrutinib inhibit inflammasome-mediated production of IL-1β, as observed in a model of influenza-induced lung injury (27). SARS-CoV-2, severe acute respiratory syndrome coronavirus 2; COVID-19, coronavirus disease 2019; ACE2, angiotensin-converting enzyme 2; TLR, Toll-like receptor; MyD88, myeloid differentiation primary response 88; BTK, Bruton tyrosine kinase; NF-κB, nuclear factor kappa B; NLRP3, NLR family pyrin domain containing 3; ASC, Apoptosis-associated speck-like protein containing a caspase recruitment domain; ORF3a, open reading frame 3a; IFN-γ, interferon gamma; IL, interleukin; IL-12R, IL-12 receptor; CCL2, C-C motif chemokine ligand 2; CXCL1, C-X-C motif chemokine ligand 1; CXCR2, C-X-C motif chemokine receptor 2.
Based on these considerations, we hypothesized that dysregulated BTK-dependent macrophage signaling is central to the exaggerated inflammatory responses and pulmonary sequelae of infection with SARS-CoV-2 and potentially other single-stranded RNA viruses. In an effort to reduce inflammation and improve clinical outcome of patients with severe COVID-19, we administered acalabrutinib, a highly specific covalent inhibitor of BTK approved in the United States for the treatment of lymphoid malignancies (28). Herein, we present a prospective off-label clinical study of 19 hospitalized patients with COVID-19 and severe hypoxia who also had evidence of inflammation and/or severe lymphopenia.
RESULTS
Patient Characteristics
This prospective off-label clinical study includes 19 hospitalized patients with severe COVID-19 who received off-label acalabrutinib between March 20, 2020 (date of treatment of the first patient) through April 10, 2020 with formal data collection completed on April 23, 2020 (Table S1). Entry criteria for this study were confirmed COVID-19 requiring hospitalization for hypoxemia (room air blood oxygen saturation (SpO2) of 94% or less) and evidence of inflammation (C-reactive protein (CRP) > 10 mg/dL and/or ferritin > 500 ng/mL) and/or lymphopenia (absolute lymphocyte count (ALC) < 1000 cells/μL). Among these patients, 13 (68%) were men and the median age was 61 years with a range of 45 to 84 years (Table 1). Eleven (58%) patients were receiving supplemental oxygen for a median of 2 days (range: 1-12), 7/11 (64%) of whom were on high flow nasal cannula at the time they began acalabrutinib (“supplemental oxygen cohort”). All but one patient had an increasing oxygen demand at the time of treatment initiation. In addition, 8 (42%) patients were receiving invasive mechanical ventilation for a median of 1.5 (range: 1-22) days prior to acalabrutinib administration (“mechanical ventilation cohort”). Coexisting medical conditions included hypertension in 16/19 (84%), obesity (body mass index > 30 kg/m2) in 13/19 (68%), and diabetes mellitus in 7/19 (37%) with a median (range) of 2 (0-5) co-morbid conditions per patient.
Table 1 Characteristics of the Patients.
Obesity is defined as body mass index ≥ 30 kg/m2, morbid obesity is defined as body mass index ≥ 40 kg/m2.
In the supplemental oxygen cohort, concomitant drugs for the treatment of COVID-19 included steroids and/or hydroxychloroquine in 5/11 (45%) patients each, and in the mechanical ventilation cohort, 6/8 (75%) and 3/8 (38%) patients, respectively, received these drugs. No patients received an anti-IL-6 receptor monoclonal antibody or remdesivir.
Laboratory evidence of inflammation with elevated CRP and/or ferritin was present in 18/19 (95%) patients with significantly elevated baseline laboratory abnormalities prior to acalabrutinib dosing including elevated CRP (> 10 mg/dL) in 15/19 (79%) patients [median (range) of 18.7 (2-31.5)]; ferritin (> 500 ng/mL) in 16/19 (84%) patients [median (range) 1240 (155-4168)]; fibrinogen (> 400 mg/dl) in 10/10 (100%) patients [median (range) 605 (409- >1000)]; D-dimer (> 0.5 mcg/mL) in 15/17 (88%) patients [median (range) 1.65 (0.48- >20)]; IL-6 (≥ 15 pg/mL) in 9/9 (100%) patients [median (range) 44 (25-89.8)]; and severely decreased ALC (≤ 1000 cells/μL) in 15/18 (83%) patients [median (range) 675 (250-1700)] (Table 1). A patient with untreated chronic lymphocytic leukemia (Patient 11) was excluded from the analysis of lymphocytes.
Oxygen Requirements During Treatment
To provide an estimate of a patient’s oxygen requirement, given different supplemental oxygen flow rates and concentrations, we computed the ratio of the percent blood oxygen saturation to the concentration of delivered oxygen (SpO2/FiO2) with higher values representing an improved oxygen uptake efficiency (Fig. 2, 3). The oxygen delivery rate, method of administration, delivered oxygen concentration (FiO2) and oxygen saturation values (SpO2/FiO2) are provided in Tables S2, S3.
Shown are measures of oxygen uptake requirement SpO2/FiO2 (% blood oxygen saturation (SpO2)/fraction of delivered oxygen (FiO2)), a ratio that accounts for both oxygen delivery and uptake (theoretical maximum 476 for 100% oxygen saturation on room air). Also shown are measures of inflammation (C-reactive protein mg/dL) and absolute lymphocyte count (cells/μL) at all available timepoints before and after acalabrutinib treatment, which was started on day 1 (dotted line). Notable clinical parameters are shown as indicated (extubation, breathing on room air, transfer to rehabilitation, hospital discharge, death). The duration of mechanical ventilation (Vent.) is indicated.
” data-hide-link-title=”0″ data-icon-position=”” href=”https://immunology.sciencemag.org/content/immunology/5/48/eabd0110/F2.large.jpg?width=800&height=600&carousel=1″ rel=”gallery-fragment-images-1266750347″ title=”Clinical course and changes in inflammatory markers during acalabrutinib treatment in patients treated prior to intubation. Shown are measures of oxygen uptake requirement SpO2/FiO2 (% blood oxygen saturation (SpO2)/fraction of delivered oxygen (FiO2)), a ratio that accounts for both oxygen delivery and uptake (theoretical maximum 476 for 100% oxygen saturation on room air). Also shown are measures of inflammation (C-reactive protein mg/dL) and absolute lymphocyte count (cells/μL) at all available timepoints before and after acalabrutinib treatment, which was started on day 1 (dotted line). Notable clinical parameters are shown as indicated (extubation, breathing on room air, transfer to rehabilitation, hospital discharge, death). The duration of mechanical ventilation (Vent.) is indicated.”>
Shown are measures of oxygen uptake requirement SpO2/FiO2 (% blood oxygen saturation (SpO2)/fraction of delivered oxygen (FiO2)), a ratio that accounts for both oxygen delivery and uptake (theoretical maximum 476 for 100% oxygen saturation on room air). Also shown are measures of inflammation (C-reactive protein mg/dL) and absolute lymphocyte count (cells/μL) at all available timepoints before and after acalabrutinib treatment, which was started on day 1 (dotted line). Notable clinical parameters are shown as indicated (extubation, breathing on room air, transfer to rehabilitation, hospital discharge, death). The duration of mechanical ventilation (Vent.) is indicated.
See legend for Fig. 2.
” data-hide-link-title=”0″ data-icon-position=”” href=”https://immunology.sciencemag.org/content/immunology/5/48/eabd0110/F3.large.jpg?width=800&height=600&carousel=1″ rel=”gallery-fragment-images-1266750347″ title=”Clinical course and changes in inflammatory markers during acalabrutinib treatment in patients treated while on mechanical ventilation. See legend for Fig. 2.”>
See legend for Fig. 2.
Among 11 patients in the supplemental oxygen cohort, the median duration of follow-up from the initiation of acalabrutinib treatment was 12 (range 10-14) days. All but one patient received at least 10 days of acalabrutinib, which was the anticipated treatment duration. At the time of formal data collection, 8 (73%) patients no longer required supplemental oxygen and had been discharged from the hospital. Among 3 patients still requiring oxygen, one was on 4L/min by nasal cannula and one was on a ventilator, both with decreasing oxygen requirements, and one patient was on continuous positive airway pressure (CPAP) with a stable oxygen requirement (Fig. 2).
Eight patients on invasive mechanical ventilation were followed for a median of 12 days (range 7-30) from the initiation of acalabrutinib treatment and received the anticipated treatment duration of 10 to 14 days, with the exception of 2 patients who died (Fig. 3). In this patient group, 4/8 (50%) were extubated, two of whom were discharged, one was on 4 L/min of oxygen and weaning, and one died of an acute pulmonary embolism. Four patients remained intubated and included three patients with oscillating oxygen requirements, and one patient who died after withdrawal of support. Two of these ventilated patients had organ dysfunction due to sepsis and renal failure.
While these results are based on our prespecified cutoff for full data analysis of April 23, 2020, we obtained an outcome update as of May 28, 2020 to assess if any patients had a disease recurrence off of acalabrutinib. In the supplementary oxygen cohort, 9 patients had been discharged on room air and remained clinically well, one was still hospitalized, and one died. In the mechanical ventilation cohort, 3 patients were discharged on room air and remained well, one was discharged to rehabilitation, and 4 patients died. In all, 12 patients achieved normal oxygenation on room air (2 more that at our formal data cutoff) and none have had a recurrence.