
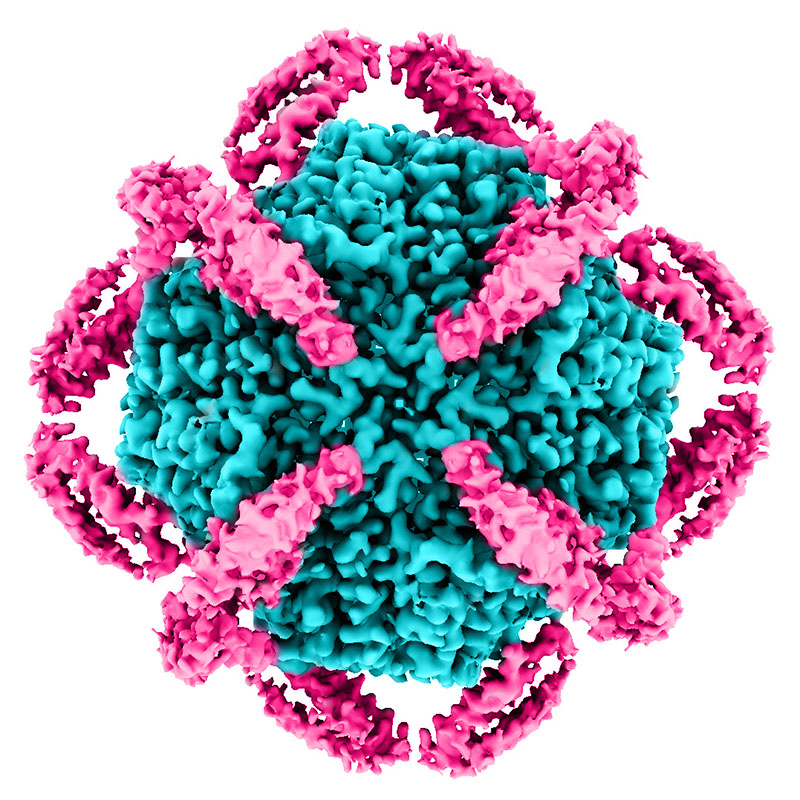
A detailed cryo-EM reconstruction of KIX proteins (magenta) surrounding the central apoferritin shell (cyan). The outer MBP shell is not shown. Credit: Greg Stewart/SLAC National Accelerator Laboratory
Sandwiching wiggly proteins between two other layers allows scientists to get the most detailed images yet of a protein that’s key to the spread of acute myeloid leukemia.
According to the American Cancer Institute, acute myeloid leukemia (AML) will afflict more than 20,000 Americans this year, killing more than 11,000 of them. Many people who are treated with intensive chemotherapy or stem cell transplants can experience side effects such as infections, hair loss, and vomiting, as well as long-term complications.
Kathleen Sakamoto, a professor at Stanford School of Medicine, has been working on the development of medicines for AML and other blood disorders in an attempt to address this situation. However, her team’s search for new ways to treat AML has been hampered by a subtle gap between two technologies used to understand the structure and function of proteins – X-ray crystallography on the one hand, and cryogenic electron microscopy (cryo-EM) on the other.
Cryo-EM reconstructions of KIX (red) sandwiched between an MBP outer shell (purple) and an apoferritin inner shell (blue). The sandwiching technique helped researchers get the best look yet of KIX, a potential target for treating acute myeloid leukemia. Credit: Greg Stewart/SLAC National Accelerator Laboratory
Researchers from Stanford University’s Schools of Medicine and Engineering and the Department of Energy’s SLAC National Accelerator Laboratory have discovered a way to close that gap by using a molecular cage to stabilize certain medium-sized proteins, allowing them to be imaged for the first time with cryo-EM, which can reveal almost atomic-level details. First authors Kaiming Zhang, a Stanford postdoctoral fellow, and Naoki Horikoshi, a visiting assistant professor, at the time of the research, and his colleagues published their results recently in the journal ACS Central Science.
At issue, says SLAC and Stanford professor Soichi Wakatsuki, is KIX, a part of the CREB Binding Protein (CBP) that AML cancer cells use to transcribe genes important for growth and survival. If researchers understood its structure better, they could design drugs that inhibit KIX and prevent cancer cells from replicating. But efforts to study the protein using X-ray crystallography haven’t been successful: The molecule’s relatively large size – by crystallography standards – makes it harder to crystallize, and even when it has been crystallized, the particulars of that process have made it harder to analyze the parts of KIX that drug designers would like to target.
At the same time, KIX is a bit too small on its own to study effectively with cryo-EM. To get good images of a protein with cryo-EM, Wakatsuki explains, you have to be able to locate many copies of the protein within an electron microscope image, then figure out how they’re oriented – whether they’re bent this way or that, and so on. Only by finding and lining up many images of a protein can cryo-EM methods yield high-resolution structures. KIX’s relatively small size – by cryo-EM standards – makes that a challenge. Another option, nuclear magnetic resonance, has been used to determine the structure of KIX when bound to other naturally occurring molecules, but the method requires extensive preparation and analysis – making it less than ideal for quickly determining molecule’s structures and therefore for less than ideal for studying the effects of potential KIX-inhibiting drugs.
The solution came to Wakatsuki and Zhang – who was working in SLAC and Stanford Professor Wah Chiu’s lab – over lunch in Tokyo, where they were working on a separate project: They would sandwich batches of KIX proteins between a central, ball-shaped molecule and an outer molecular cage. Because this “double shell” was much larger than individual KIX molecules, it would be easier to spot and orient in cryo-EM images, and that would make it easier to get high-resolution images of the KIX molecules themselves.
In addition to seeing KIX’s structure, Wakatsuki said, his lab and Chiu’s worked with Sakamoto and Stanford computer science professor Ron Dror and were able to add other molecules to the mix to see if they might bind to and potentially inhibit KIX’s function. Already, the team reports, they’ve been able to make that bonding about 200 times stronger, which could help scientists develop drugs that are effective at lower doses. “The name of the game is to find compounds that inhibit KIX at lower concentrations,” Wakatsuki said. “This is still not good enough, but we have made progress.”
The team’s results also suggest this method could prove useful for other proteins of in-between sizes that are hard to study with either cryo-EM or X-ray crystallography – including, perhaps, some viral proteins. “We are moving forward to expand the applicability of the approach,” Wakatsuki said.
The research was funded by the National Institutes of Health, the Pediatric Cancer Research Foundation, Maternal Child Health Research Institute, Stanford University, and the Leukemia and Lymphoma Society. Zhang received startup funding from the University of Science and Technology of China.
Reference: “Cryo-EM, Protein Engineering, and Simulation Enable the Development of Peptide Therapeutics against Acute Myeloid Leukemia” by Kaiming Zhang, Naoki Horikoshi, Shanshan Li, Alexander S. Powers, Mikhail A. Hameedi, Grigore D. Pintilie, Hee-Don Chae, Yousuf A. Khan, Carl-Mikael Suomivuori, Ron O. Dror, Kathleen M. Sakamoto, Wah Chiu and Soichi Wakatsuki, 7 February 2022, ACS Central Science.
DOI: 10.1021/acscentsci.1c01090