
Summary
Background
This is the first randomised controlled trial for assessment of the immunogenicity and safety of a candidate non-replicating adenovirus type-5 (Ad5)-vectored COVID-19 vaccine, aiming to determine an appropriate dose of the candidate vaccine for an efficacy study.
Methods
This randomised, double-blind, placebo-controlled, phase 2 trial of the Ad5-vectored COVID-19 vaccine was done in a single centre in Wuhan, China. Healthy adults aged 18 years or older, who were HIV-negative and previous severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection-free, were eligible to participate and were randomly assigned to receive the vaccine at a dose of 1 × 1011 viral particles per mL or 5 × 1010 viral particles per mL, or placebo. Investigators allocated participants at a ratio of 2:1:1 to receive a single injection intramuscularly in the arm. The randomisation list (block size 4) was generated by an independent statistician. Participants, investigators, and staff undertaking laboratory analyses were masked to group allocation. The primary endpoints for immunogenicity were the geometric mean titres (GMTs) of specific ELISA antibody responses to the receptor binding domain (RBD) and neutralising antibody responses at day 28. The primary endpoint for safety evaluation was the incidence of adverse reactions within 14 days. All recruited participants who received at least one dose were included in the primary and safety analyses. This study is registered with ClinicalTrials.gov, NCT04341389.
Findings
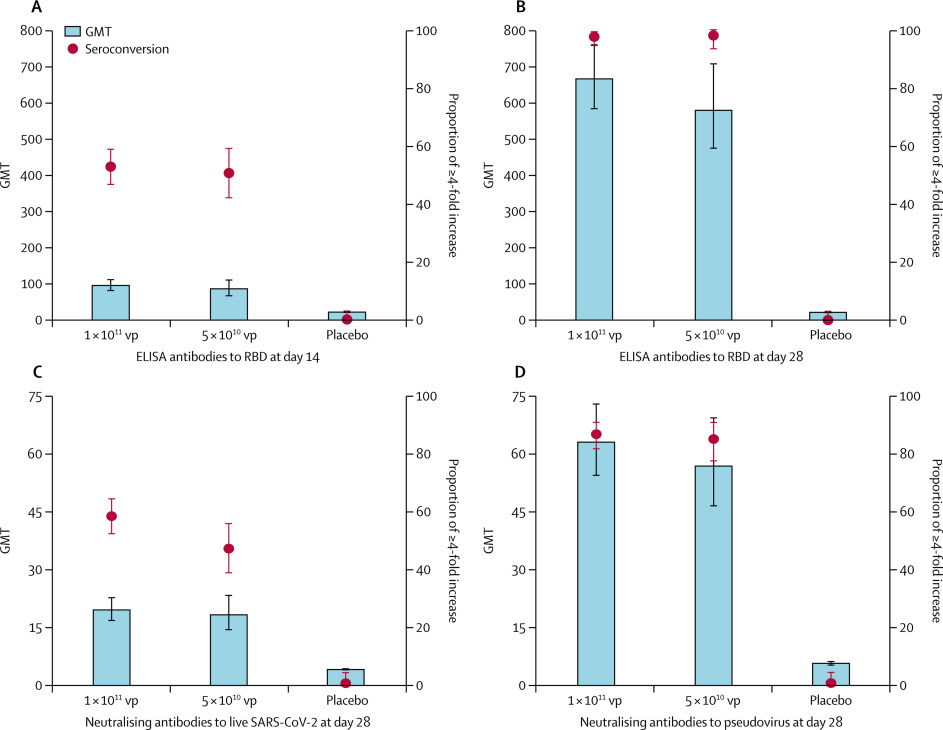
603 volunteers were recruited and screened for eligibility between April 11 and 16, 2020. 508 eligible participants (50% male; mean age 39·7 years, SD 12·5) consented to participate in the trial and were randomly assigned to receive the vaccine (1 × 1011 viral particles n=253; 5 × 1010 viral particles n=129) or placebo (n=126). In the 1 × 1011 and 5 × 1010 viral particles dose groups, the RBD-specific ELISA antibodies peaked at 656·5 (95% CI 575·2–749·2) and 571·0 (467·6–697·3), with seroconversion rates at 96% (95% CI 93–98) and 97% (92–99), respectively, at day 28. Both doses of the vaccine induced significant neutralising antibody responses to live SARS-CoV-2, with GMTs of 19·5 (95% CI 16·8–22·7) and 18·3 (14·4–23·3) in participants receiving 1 × 1011 and 5 × 1010 viral particles, respectively. Specific interferon γ enzyme-linked immunospot assay responses post vaccination were observed in 227 (90%, 95% CI 85–93) of 253 and 113 (88%, 81–92) of 129 participants in the 1 × 1011 and 5 × 1010 viral particles dose groups, respectively. Solicited adverse reactions were reported by 183 (72%) of 253 and 96 (74%) of 129 participants in the 1 × 1011 and 5 × 1010 viral particles dose groups, respectively. Severe adverse reactions were reported by 24 (9%) participants in the 1 × 1011 viral particles dose group and one (1%) participant in the 5 × 1010 viral particles dose group. No serious adverse reactions were documented.
Interpretation
The Ad5-vectored COVID-19 vaccine at 5 × 1010 viral particles is safe, and induced significant immune responses in the majority of recipients after a single immunisation.
Funding
National Key R&D Programme of China, National Science and Technology Major Project, and CanSino Biologics.
Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has caused more than 12·1 million cases of COVID-19 worldwide, resulting in 551 000 deaths and severe economic disruption.
WHO
Coronavirus disease (COVID-2019) situation reports.
,
- Graham BS
Rapid COVID-19 vaccine development.
After the initial outbreak, with more than 80 000 cases and 3000 deaths in China, COVID-19 has now spread to 216 countries and territories. Large numbers of cases and deaths are reported daily from Europe, the USA, Brazil, Russia, India, and many other countries.
- Cheng HY
- Jian SW
- Liu DP
- Ng TC
- Huang WT
- Lin HH
Contact tracing assessment of COVID-19 transmission dynamics in Taiwan and risk at different exposure periods before and after symptom onset.
,
- Leung K
- Wu JT
- Liu D
- Leung GM
First-wave COVID-19 transmissibility and severity in China outside Hubei after control measures, and second-wave scenario planning: a modelling impact assessment.
The current pandemic has highlighted the need for effective preventive solutions to reduce burden and spread of the disease. As long as there is a COVID-19 epidemic in one area in the world, there is a risk of a pandemic.
Research in context
Evidence before this study
We searched PubMed on July 16, 2020, for clinical trial reports with the terms “COVID-19” or “SARS-CoV-2”, “vaccine”, and “clinical trial”. Using the same terms, we also searched ClinicalTrials.gov for unpublished trials of COVID-19 vaccines. Except for the results of our earlier phase 1 study with the adenovirus type-5 (Ad5)-vectored vaccine and a phase 1 clinical trial with an mRNA vaccine (mRNA-1273) done in a small number of participants, no other COVID-19 vaccine data from clinical trials have been reported. We found registered trials with 11 candidate COVID-19 vaccines at ClinicalTrials.gov, including three recombinant protein-based vaccines, two viral vector-based vaccines, one DNA vaccine, two mRNA vaccines, two inactivated virus vaccines, and one autologous dendritic cell-based vaccine loaded with antigens from severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). The majority of the trials registered were in early phases; only ChAdOx1 nCoV-19 developed by the University of Oxford (Oxford, UK) is going to be evaluated in a phase 3 trial.
In the previously reported open-label, non-randomised, phase 1 trial, we found that the Ad5-vectored COVID-19 vaccine was tolerable and immunogenic in healthy adults. One dose of the vaccine induced rapid specific T-cell and humoral responses by 14 days.
Added value of this study
This study provides more evidence for the immunogenicity and safety of the Ad5-vectored COVID-19 vaccine in a larger population. To assess the vaccine in a more diverse population, we removed the age cap for the recruitment of participants for this phase 2 trial. Older individuals (ie, aged ≥55 years), many of whom often have chronic illness, have a high risk of serious illness and death associated with SARS-CoV-2 infection; thus, they are an important target population for a COVID-19 vaccine. Our results suggest a single-dose immunisation schedule of Ad5-vectored COVID-19 vaccine at 5 × 1010 viral particles is an appropriate regimen for healthy adults. Compared with the younger population, we found older people to have a significantly lower immune response, but higher tolerability, to the Ad5-vectored COVID-19 vaccine. Therefore, an additional dose might be needed to induce a better immune response in the older population, and this will be evaluated in a phase 2b trial.
Implications of all the available evidence
Evidence from this phase 2 study indicates the candidate Ad5-vectored COVID-19 vaccine has a good safety profile, with only mild, transient adverse events related to vaccination and no serious adverse events. Single-dose immunisation with the vaccine induced rapid onset of immune responses within 14 days and significant humoral and cellular immune responses within 28 days in the majority of the recipients. We are planning an international multicentre, randomised, double-blind, controlled phase 3 effectiveness trial to further evaluate the efficacy of the vaccine. We are in the midst of a global COVID-19 pandemic; thus, timely sharing of the results of clinical trials with candidate vaccines is critical.
Unlike typical vaccine development, which often takes decades, developing a vaccine to prevent COVID-19 has become a race between humans and the virus.
- Lurie N
- Saville M
- Hatchett R
- Halton J
Developing COVID-19 vaccines at pandemic speed.
Many countries have accelerated the process of clinical trials to determine an effective and safe vaccine to prevent COVID-19 and influence the course of the current pandemic.
- Caddy S
Developing a vaccine for covid-19.
,
WHO
Draft landscape of COVID-19 candidate vaccines.
Currently, about 250 candidate vaccines against SARS-CoV-2 are in development worldwide, including mRNA vaccines, replicating or non-replicating viral vectored vaccines, DNA vaccines, autologous dendritic cell-based vaccine, and inactive virus vaccines.
CEPI
CEPI-funded COVID-19 vaccine candidates progress to clinical trials.
To date, at least 17 of these vaccine candidates are under evaluation in clinical trials.
In March 2020, we did a single-centre, open-label, non-randomised, first-in-human phase 1 trial
Zhu FC, Li YH, Guan XH, et al. Safety, tolerability, and immunogenicity of a recombinant adenovirus type-5 vectored COVID-19 vaccine: a dose-escalation, open-label, non-randomised, first-in-human trial. 395: 1845–54.
with CanSino Biologics’ (Tianjin, China) adenovirus type-5 (Ad5)-vectored COVID-19 vaccine in a dose-escalating manner (5 × 1010, 1 × 1011, and 1·5 × 1011 viral particles). Generally, the candidate vaccines had acceptable safety and tolerability profiles and promising immunogenicity in healthy Chinese adults; however, the high-dose vaccine was associated with an increased risk of severe adverse reactions. We therefore carried forward the phase 2 trial with only the 5 × 1010 and 1 × 1011 viral particles doses of the candidate vaccine, aiming to further evaluate the immunogenicity and safety in a larger population, and to determine an appropriate dose for the efficacy study.
Methods
Study design and participants
This randomised, double-blind, placebo-controlled, phase 2 trial of the Ad5-vectored COVID-19 vaccine was done in a single centre in Wuhan (Hubei province, China). The study was done in accordance with the Declaration of Helsinki and Good Clinical Practice. An independent data safety monitoring board was established before the start of the trial to provide oversight of the safety data during the study. The trial protocol was reviewed and approved by the National Medical Products Administration, China, and the institutional review board of the Jiangsu Provincial Center of Disease Control and Prevention. The protocol is available online.
Eligible participants were healthy adults aged 18 years or older, who were HIV-negative and previous SARS-CoV-2 infection-free, confirmed by commercial human immunodeficiency virus antibody detection kit (InTec products, Xiamen, China) and SARS-CoV-2 rapid test kit (Jinwofu, Beijing, China) using fingertip blood at screening. To be included, participants needed to be able to understand the content of informed consent and willing to sign the informed consent; able and willing to complete all the scheduled study processes; have an axillary temperature of 37·0°C or less; have a body-mass index of between 18·5 and 30·0; and have general good health as established by medical history and physical examination. Pregnant or breastfeeding women were excluded. People with mental disease, history of allergies, or serious cardiovascular disease, and some other major chronic illnesses were also excluded. A complete list of the inclusion and exclusion criteria is provided in the protocol. Participants were recruited through online recruitment advertisements. Written informed consent was obtained from each participant before screening for eligibility.
Randomisation and masking
The Ad5-vectored COVID-19 vaccine was developed by the Beijing Institute of Biotechnology (Beijing, China) and CanSino Biologics, and contained replication-defective Ad5 vectors expressing the full-length spike gene based on Wuhan-Hu-1 (GenBank accession number YP_009724390). The placebo contained the vaccine excipients only, with no viral particles. The experimental vaccines and placebos had identical packaging with a randomisation number on each vial as the only identifiers. The vaccines of 1 × 1011 and 5 × 1010 viral particles and placebo were randomised at a 2:1:1 ratio. Eligible participants were sequentially assigned a randomisation number, according to a blocked randomisation list (block size 4) generated by an independent statistician using SAS software (version 9.4), and injected with an experimental vaccine or placebo labelled with the same number. Individuals involved in randomisation and masking had no involvement in the rest of the trial. Participants, investigators, and staff undertaking laboratory analyses were masked to group allocation.
Procedures
A single injection of the vaccines of 1 × 1011 or 5 × 1010 viral particles per mL, or placebo, were given to participants intramuscularly in the arm. Participants were monitored for 30 min post injection for immediate adverse reactions, and followed up for any injection site or systemic adverse reactions within 14 days and adverse events within 28 days post vaccination. Serious adverse events self-reported by participants were documented throughout the study.
The detailed methods of the assays have been reported previously.
Zhu FC, Li YH, Guan XH, et al. Safety, tolerability, and immunogenicity of a recombinant adenovirus type-5 vectored COVID-19 vaccine: a dose-escalation, open-label, non-randomised, first-in-human trial. 395: 1845–54.
In brief, blood samples were taken from participants at day 0 immediately before vaccination, and at days 14 and 28 post vaccination for the measurement of specific antibody responses against the receptor binding domain (RBD) using ELISA kits (Beijing Wantai BioPharm, Beijing, China). The detection limit for the RBD-specific ELISA antibody test was 1: 40. The neutralising antibody responses to live SARS-CoV-2 virus (strain SARS-CoV-2/human/CHN/Wuhan_IME-BJ01/2020, GenBank number MT291831.1) or a pseudovirus (a vesicular stomatitis virus pseudovirus system expressing the spike glycoprotein),
- Nie J
- Li Q
- Wu J
- et al.
Establishment and validation of a pseudovirus neutralization assay for SARS-CoV-2.
and cellular immune responses before the vaccination and 28 days after the vaccination were also measured. The detection limits for the neutralising antibody tests to live SARS-CoV-2 virus and a pseudovirus were 1:8 and 1: 10, respectively. Undetectable antibody titres in serum were assigned values of half the detection limits for calculation. The cellular immune responses of the expression of interferon (IFN) γ stimulated by the overlapping peptide pool of spike glycoprotein were detected by enzyme-linked immunospot (ELISpot) assay (Mabtech, Stockholm, Sweden). Positive IFNγ-ELISpot response was defined as at least five spot-forming cells per 1 × 105 peripheral blood mononuclear cells, and a minimum of a two-times increase from baseline. Neutralising antibody titres against the vaccine vector Ad5 were measured with the serum neutralisation assay.
- Sprangers MC
- Lakhai W
- Koudstaal W
- et al.
Quantifying adenovirus-neutralizing antibodies by luciferase transgene detection: addressing preexisting immunity to vaccine and gene therapy vectors.
The follow-ups were scheduled at days 14 and 28, and month 6 post vaccination for safety and immunogenicity assessment.
Outcomes
The primary objectives were to evaluate immunogenicity and safety of the Ad5-vectored COVID-19 vaccine, and to determine a vaccine dose for a phase 3 efficacy study. The primary endpoint for safety evaluation was the incidence of adverse reactions within 14 days after the injection. The primary endpoints for immunogenicity were the geometric mean titres (GMTs) of RBD-specific ELISA antibody responses and neutralising antibody responses against live virus or pseudovirus at day 28 post vaccination. The secondary endpoints for immunogenicity were RBD-specific ELISA antibody responses at day 14 and month 6 (6 month data not yet available), and specific T-cell responses at day 28 post vaccination. Seroconversion of the humoral immune responses was also a secondary endpoint, and was defined as an increase in post -vaccination titre of at least four-times from baseline. The secondary safety outcomes included the occurrence of adverse events from days 0 to 28 after vaccination, and serious adverse events reported up to 6 months. Investigators did severity grading of the adverse events according to the standard guidelines issued by the China State Food and Drug Administration, and the causality with immunisation before unmasking. Stratified analysis of safety and immunogenicity of the participants was done based on the baseline Ad5 neutralising antibody titres with a cutoff at 1: 200. Post-hoc analysis of the immune responses by age and sex, and the proportion of vaccine recipients with the composite endpoint of either positive cellular or seroconversion of humoral immune responses at day 28 post vaccination were presented.
Statistical analysis
This phase 2 trial was launched before the immunogenicity data from the phase 1 trial were obtained; therefore, the sample size was not calculated at the design stage. An overall sample size of 500 participants (n=250 in the 1 × 1011 viral particles dose group; n=125 in the 5 × 1010 viral particles dose group; and n=125 in the placebo group) was determined, based on expert opinion and the minimum sample size requirement in the technical guidelines for vaccine clinical trials issued by the National Medical Products Administration, China.
National Medical Products Administration, China
Technical guidelines for clinical trials of vaccines.
We did an ex-post power calculation of this study after the immunogenicity data from the phase 1 study were available, using PASS software (version 11.0). For the RBD-specific antibody, 250 individuals in the 1 × 1011 viral particles dose group and 125 individuals in the 5 × 1010 viral particles dose group were able to provide at least a power of 90% to show a difference of the log-transferred titre of 0·176 with an SD of 0·4 between the dose groups, at a level of significance of 0·017, considering multiple comparisons.
Statistical tests were two-sided with an α value of 0·05, and analysed by an independent statistician using SAS (version 9.4). The primary immunogenicity analysis was done in the full-analysis cohort, including all participants who were injected and donated blood samples for antibody tests post vaccination, while the safety analysis was done in all enrolled participants who received the vaccination. Correlation analysis of the RBD-specific ELISA antibody and neutralising antibody was done, and the Pearson correlation coefficient was calculated. Antibody responses are reported as the GMT with 95% CI. ANOVA was used for log-transformed antibody titres, and the Wilcoxon rank-sum test for data that were not normally distributed. The χ2 test or Fisher’s exact test was used for categorical data. Multiple comparisons were done if a significant difference across the treatment groups was noted, using Student Newman-Keuls test or Bonferroni-adjusted α value when relevant. This trial is registered with ClinicalTrials.gov, NCT04341389.
Role of the funding source
The funders of the study were involved in protocol design, but not in data collection, statistical analysis, data interpretation, or writing of the report. All the authors had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Results
603 volunteers were recruited and screened for eligibility between April 11 and 16, 2020 (figure 1). 95 individuals were excluded, leaving 508 eligible participants who consented to participate in the trial and were randomly assigned to vaccine or placebo. 253 were randomly assigned to the 1 × 1011 viral particles dose group, 129 to the 5 × 1010 viral particles dose group, and 126 to the placebo group. The mean age of the participants was 39·7 years (SD 12·5; range 18–83), with 309 (61%) individuals aged 18–44 years, 134 (26%) aged 45–54 years, and 65 (13%) aged 55 years or older across the treatment groups (table 1). 254 (50%) of 508 participants were male. Baseline characteristics of the participants and the pre-existing Ad5 neutralising antibody titres were largely similar across the treatment groups. Among the 508 participants, 266 (52%) had high pre-existing immunity and 242 (48%) had low pre-existing immunity to the Ad5 vector. All the participants completed the scheduled safety visits within 28 days post vaccination and gave blood samples at days 0 and 28, and 506 (>99%) donated blood samples at day 14.
Table 1Baseline characteristics
Data are number of participants (%) or mean (SD). vp=viral particles.
The baseline antibody titres of the participants are given in the appendix (p 1). RBD-specific ELISA antibody responses induced by the Ad5-vectored COVID-19 vaccine were detected from day 14 onwards, with GMTs of 94·5 (95% CI 80·5–110·8) and 85·1 (66·0–109·7) in the 1 × 1011 and 5 × 1010 viral particles dose groups, respectively (figure 2). At day 28, the RBD-specific ELISA antibodies peaked at 656·5 (575·2–749·2) in the 1 × 1011 viral particles dose group and 571·0 (467·6–697·3) in the 5 × 1010 viral particles dose group. 244 (96%, 95% CI 93–98) of 253 participants in the 1 × 1011 viral particles dose group and 125 (97%, 92–99) of 129 participants in the 5 × 1010 viral particles dose group showed seroconversion of RBD-specific ELISA antibodies at day 28, whereas the participants in the placebo group showed no antibody increase from baseline.

Figure 2Specific antibody responses to RBD, neutralising antibodies to live severe acute respiratory syndrome coronavirus 2 and pseudovirus post vaccination
Seroconversion was defined as an increase in post-vaccination titre of at least four-times baseline. The baseline antibody titres are shown in the appendix (p 1). All comparisons across the three treatment groups are p<0·0001. Multiple comparisons showed no significant difference between the 1 × 1011 vp a